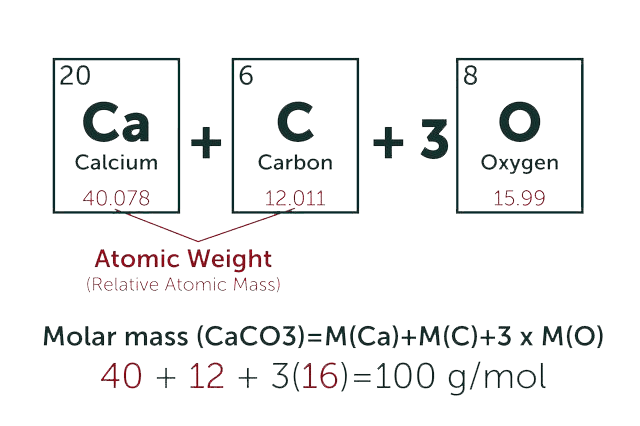
Cercosporamide
* Please be kindly noted products are not for therapeutic use. We do not sell to patients.

| Category | Enzyme inhibitors |
| Catalog number | BBF-04578 |
| CAS | 131436-22-1 |
| Molecular Weight | 331.28 |
| Molecular Formula | C16H13NO7 |
| Purity | >95% |
Online Inquiry
Description
Cercosporamide, a natural antifungal phytotoxin produced by Cercosporidium sp. MST-FP1899, has been found to be a selective Mnk inhibitor and shows probable antileukemic effects.
Specification
| Synonyms | (9aS)-8-acetyl-1,3,7-trihydroxy-9a-methyl-9-oxo-9,9a-dihydrodibenzo[b,d]furan-4-carboxamide; (S)-8-Acetyl-9,9a-dihydro-1,3,7-trihydroxy-9a-methyl-9-oxo-4-dibenzofurancarboxamide; (-)-Cercosporamide |
| Shelf Life | As supplied, 2 years from the QC date provided on the Certificate of Analysis, when stored properly |
| Storage | Store at -20°C |
| IUPAC Name | (9aS)-8-acetyl-1,3,7-trihydroxy-9a-methyl-9-oxodibenzofuran-4-carboxamide |
| Canonical SMILES | CC(=O)C1=C(C=C2C(C1=O)(C3=C(C=C(C(=C3O2)C(=O)N)O)O)C)O |
| InChI | InChI=1S/C16H13NO7/c1-5(18)10-7(20)4-9-16(2,14(10)22)12-8(21)3-6(19)11(15(17)23)13(12)24-9/h3-4,19-21H,1-2H3,(H2,17,23)/t16-/m1/s1 |
| InChI Key | GEWLYFZWVLXQME-MRXNPFEDSA-N |
| Source | Cercosporidium sp. |
Properties
| Appearance | Light to Dark Tan Solid |
| Application | Insecticide |
| Antibiotic Activity Spectrum | Fungi |
| Boiling Point | 582.5°C at 760 mmHg |
| Melting Point | 188-189°C |
| Density | 1.7 g/cm3 |
| Solubility | Soluble in DMSO (10 mM), Chloroform (1 mg/mL) |
Toxicity
| Carcinogenicity | No indication of carcinogenicity (not listed by IARC). |
| Mechanism Of Toxicity | Dibenzofurans bind the aryl hydrocarbon receptor, which increases its ability to activate transcription in the XRE promoter region. This alters the expression of a number of genes. |
Reference Reading
1.Synthesis and antiproliferative activity of benzofuran-based analogs of cercosporamide against non-small cell lung cancer cell lines.
Bazin MA1, Bodero L, Tomasoni C, Rousseau B, Roussakis C, Marchand P. Eur J Med Chem. 2013 Nov;69:823-32. doi: 10.1016/j.ejmech.2013.09.013. Epub 2013 Sep 18.
A novel series of 3-methyl-1-benzofuran derivatives were synthesized and screened in vitro for their antiproliferative activity against two human NSCLC cell lines (NSCLC-N6 mutant p53 and A549 wild type p53). Most promising compounds presented a structural analogy with the west part of cercosporamide, a natural product of biological interest. In particular, compounds 10, 12 and 31 showed cytotoxic activities at micromolar concentrations (IC₅₀ < 9.3 μM) and compounds 13, 18 and 32 displayed moderate IC₅₀ values (25-40 μM).
2.Discovery of a BTK/MNK dual inhibitor for lymphoma and leukemia.
Wu H1,2, Hu C1, Wang A1,2, Weisberg EL3, Chen Y1, Yun CH4, Wang W1, Liu Y3, Liu X1,2, Tian B5, Wang J4, Zhao Z1, Liang Y4, Li B1, Wang L1, Wang B1, Chen C1, Buhrlage SJ4, Qi Z1, Zou F1, Nonami A3, Li Y3, Fernandes SM3, Adamia S3, Stone RM3, Galinsky IA3, Leukemia. 2016 Jan;30(1):173-81. doi: 10.1038/leu.2015.180. Epub 2015 Jul 13.
Bruton's tyrosine kinase (BTK) kinase is a member of the TEC kinase family and is a key regulator of the B-cell receptor (BCR)-mediated signaling pathway. It is important for B-cell maturation, proliferation, survival and metastasis. Pharmacological inhibition of BTK is clinically effective against a variety of B-cell malignances, such as mantle cell lymphoma, chronic lymphocytic leukemia (CLL), acute myeloid leukemia (AML) and activated B-cell-diffuse large B-cell lymphoma. MNK kinase is one of the key downstream regulators in the RAF-MEK-ERK signaling pathway and controls protein synthesis via regulating the activity of eIF4E. Inhibition of MNK activity has been observed to moderately inhibit the proliferation of AML cells. Through a structure-based drug-design approach, we have discovered a selective and potent BTK/MNK dual kinase inhibitor (QL-X-138), which exhibits covalent binding to BTK and noncovalent binding to MNK. Compared with the BTK kinase inhibitor (PCI-32765) and the MNK kinase inhibitor (cercosporamide), QL-X-138 enhanced the antiproliferative efficacies in vitro against a variety of B-cell cancer cell lines, as well as AML and CLL primary patient cells, which respond moderately to BTK inhibitor in vitro.
3.First MNKs degrading agents block phosphorylation of eIF4E, induce apoptosis, inhibit cell growth, migration and invasion in triple negative and Her2-overexpressing breast cancer cell lines.
Ramalingam S1, Gediya L, Kwegyir-Afful AK, Ramamurthy VP, Purushottamachar P, Mbatia H, Njar VC. Oncotarget. 2014 Jan 30;5(2):530-43.
Some retinoic acid metabolism blocking agents (RAMBAs) are known to exhibit a wide range of anticancer activities by mechanisms that are still not completely resolved. This study investigated the anticancer efficacy and mechanism(s) of novel RAMBA retinamides (RRs) in triple negative and Her-2 overexpressing breast cancer cells. Specifically, we examined the possibility that RRs affect the translational machinery in these breast cancer (BC) cells. Recent findings suggest that overexpression of eukaryotic translation initiation factor 4E (eIF4E) in breast cancers critically augments CAP-dependent mRNA translation and synthesis of proteins involved in cell growth, cell proliferation, invasion and apoptosis evasion. The oncogenic potential of eIF4E is strictly dependent on serine209 phosphorylation by upstream MAPK-interacting kinases (Mnks). Targeting Mnk/eIF4E pathway for blocking Mnk function and eIF4E phosphorylation is therefore a novel approach for treating BCs, particularly for Her2-positive and triple negative breast cancers that have no indications for endocrine therapy or effective treatment regimes.
4.Pharmacology and in vitro profiling of a novel peroxisome proliferator-activated receptor γ ligand, Cerco-A.
Wakabayashi K1, Hayashi S, Matsui Y, Matsumoto T, Furukawa A, Kuroha M, Tanaka N, Inaba T, Kanda S, Tanaka J, Okuyama R, Wakimoto S, Ogata T, Araki K, Ohsumi J. Biol Pharm Bull. 2011;34(7):1094-104.
Peroxisome proliferator-activated receptor γ (PPARγ; NR1C3) is known as a key regulator of adipocytogenesis and the molecular target of thiazolidinediones (TZDs), also known as antidiabetic agents. Despite the clinical benefits of TZDs, their use is often associated with adverse effects including peripheral edema, congestive heart failure, and weight gain. Here we report the identification and characterization of a non-thiazolidinedione PPARγ partial agonist, Cerco-A, which is a derivative of the natural product, (-)-cercosporamide. Cerco-A was found to be a binder of the PPARγ ligand-binding domain in a ligand competitive binding assay and showed a unique cofactor recruitment profile compared to rosiglitazone. A crystal structure analysis revealed that Cerco-A binds to PPARγ without direct hydrogen bonding to helix12. In PPARγ transcriptional activation assay and an adipocyte differentiation assay, Cerco-A was a potent partial agonist of PPARγ.
Spectrum
Predicted GC-MS Spectrum - GC-MS (Non-derivatized) - 70eV, Positive

Experimental Conditions
Ionization Mode: Positive
Ionization Energy: 70 eV
Chromatography Type: Gas Chromatography Column (GC)
Instrument Type: Single quadrupole, spectrum predicted by CFM-ID(EI)
Mass Resolution: 0.0001 Da
Molecular Formula: C16H13NO7
Molecular Weight (Monoisotopic Mass): 331.0692 Da
Molecular Weight (Avergae Mass): 331.2769 Da
Ionization Energy: 70 eV
Chromatography Type: Gas Chromatography Column (GC)
Instrument Type: Single quadrupole, spectrum predicted by CFM-ID(EI)
Mass Resolution: 0.0001 Da
Molecular Formula: C16H13NO7
Molecular Weight (Monoisotopic Mass): 331.0692 Da
Molecular Weight (Avergae Mass): 331.2769 Da
Predicted LC-MS/MS Spectrum - 10V, Positive

Experimental Conditions
Ionization Mode: Positive
Collision Energy: 10 eV
Instrument Type: QTOF (generic), spectrum predicted by CFM-ID
Mass Resolution: 0.0001 Da
Molecular Formula: C16H13NO7
Molecular Weight (Monoisotopic Mass): 331.0692 Da
Molecular Weight (Avergae Mass): 331.2769 Da
Collision Energy: 10 eV
Instrument Type: QTOF (generic), spectrum predicted by CFM-ID
Mass Resolution: 0.0001 Da
Molecular Formula: C16H13NO7
Molecular Weight (Monoisotopic Mass): 331.0692 Da
Molecular Weight (Avergae Mass): 331.2769 Da
Recommended Products
| BBF-03754 | CASTANOSPERMINE | Inquiry |
| BBF-01829 | Deoxynojirimycin | Inquiry |
| BBF-03753 | Baicalin | Inquiry |
| BBF-01826 | Deoxymannojirimycin | Inquiry |
| BBF-02614 | Nystatin | Inquiry |
| BBF-03755 | Actinomycin D | Inquiry |
Bio Calculators
* Our calculator is based on the following equation:
Concentration (start) x Volume (start) = Concentration (final) x Volume (final)
It is commonly abbreviated as: C1V1 = C2V2
* Total Molecular Weight:
g/mol
Tip: Chemical formula is case sensitive. C22H30N4O √ c22h30n40 ╳